





































Pure Essential de BÜCHI Sistema de Cromatografía

Ideal para usuarios que dan sus primeros pasos en la cromatografía automatizada el Sistema de Cromatografía Pure Essential de BÜCHI se centra en los aspectos básicos de cualquier aplicación flash básica. Gracias al diseño del instrumento, su modularidad y su facilidad de uso, le resultará muy fácil empezar con la automatización y se beneficiará de una mayor velocidad y de infinitas posibilidades.
El Sistema de Cromatografía Pure Essential ofrece como ventajas un diseño y una configuración básicos. Es ideal como instrumento básico y para empezar con la automatización. Además del hardware, BÜCHI ha desarrollado un software que permite al usuario controlar de manera rápida y sencilla los parámetros más importantes de una ejecución. Se pueden conectar fácilmente y configurar automáticamente complementos como el detector UV o el colector de fracciones.

Solución modular y ampliable
Siempre se empieza con lo mínimo. BÜCHI le proporciona asistencia en cada paso de su proceso de investigación y le ofrece la máxima flexibilidad en su paso de purificación. El Sistema de Cromatografía Pure C-900 acelera sus procesos de separación y aumenta la reproducibilidad. No obstante, en cualquier momento puede aumentar aún más la eficiencia de sus procesos de separación añadiendo un colector de fracciones y un detector UV para disfrutar de estas ventajas: recolección de fracciones activada por volumen o UV, supervisión de la eficiencia de los procesos de separación, detección de compuestos durante una ejecución y reducción de la evaporación de disolvente gracias a una recolección más precisa.
Cromatografía Plug and Play
Con los Sistemas de Cromatografía Pure Essential, las separaciones flash son más sencillas que nunca gracias a varias características clave del hardware y el software.
El Sistema de Cromatografía Pure Essential se puede instalar manualmente de manera sencilla, y tanto el colector de fracciones como el detector UV se configuran automáticamente. Gracias al innovador software de última generación, el sistema se puede controlar de manera intuitiva. Con solo unos clics, se puede programar un método o adaptar parámetros sobre la marcha.
¿Por qué el Nuevo Sistema de Cromatografía Pure Essential de BÜCHI es ideal si me encuentro dando mis primeros pasos en Cromatografía?
El Sistema de Cromatografía Pure Essential se centra en los aspectos básicos de cualquier aplicación flash básica. Gracias al diseño del instrumento, su modularidad y su facilidad de uso, le resultará muy fácil empezar con la automatización y se beneficiará de una mayor velocidad y de infinitas posibilidades.

Tamaño compacto y solidez
Gracias a las reducidas dimensiones del Sistema de Cromatografía Pure Essential, se puede colocar incluso en laboratorios abarrotados. La bomba, que incluye el detector UV, se puede situar encima del colector de fracciones para ahorrar espacio. El dispositivo ofrece como ventajas una construcción de calidad sólida en general con cabezales de bomba de acero, racks del colector de fracciones extremadamente robustos y un resistente soporte para cartuchos. Está fabricado para resistir los rigores del uso diario.
Básico a la vez que potente y seguro
Aunque es básico, el Sistema de Cromatografía Pure Essential destaca por su potencia y seguridad. La bomba de 50 bares puede gestionar cualquier aplicación flash, con partículas de sílice grandes o pequeñas. Sus tres pistones garantizan un flujo sin pulsaciones que contribuye a una separación eficiente. El detector UV es muy sensible y permite detectar compuestos en concentraciones incluso más bajas. Además, el instrumento está equipado con un sensor de presión y es poco susceptible a los derrames gracias a su diseño inteligente.
Muchas separaciones flash solo requieren una configuración básica. Solo se necesita una bomba para agilizar el flujo de disolventes y muestras y, opcionalmente, un detector UV y un colector de fracciones.
Carga de muestras y recolección de fracciones sin límites
El Sistema de Cromatografía Pure Essential se adapta a sus necesidades.
El colector de fracciones es compatible con recipientes de diferentes tamaños y diseños: tubo, embudos y matraces. Además, el usuario puede elegir entre la carga de muestras líquidas o sólidas en función de la aplicación. Las muestras líquidas se pueden inyectar directamente mediante una válvula de inyección manual o mediante bucles o cámaras de muestras con capacidades de volumen entre 5 y 1.000 ml.
Se adapta a sus aplicaciones
Las aplicaciones pueden variar ampliamente en cuanto a escala y complejidad. Con el Sistema de Cromatografía Pure Essential, tendrá el equipo que necesita para cualquier aplicación. Puede conectar cualquier tipo de cartucho flash (con tamaños entre 4 y 5.000 g) o columnas de vidrio (con un diámetro interno de hasta 100 mm y longitudes de hasta 900 mm) en función de la cantidad de muestra que se vaya a purificar. Se puede instalar una cámara de mezclado a alta presión adicional (con tamaños entre 2,5 y 23 ml) para mezclar correctamente sus disolventes, especialmente cuando se usen en concentraciones bajas.
 Opciones adicionales
Opciones adicionales
La separación de una mezcla suele acabar con múltiples fracciones recogidas. La concentración de estas fracciones se puede llevar a cabo mediante el Rotavapor® de BÜCHI o mediante la tecnología Vortex de evaporación con el SyncorePlus de BÜCHI, que permite procesar varias muestras a la vez. El Sistema de Cromatografía Pure Essential es compatible con los mismos tubos de vidrio y racks del Rotavapor® y el SyncorePlus, lo que maximiza la eficiencia de sus procesos.
Siempre se empieza con lo mínimo. BÜCHI le proporciona asistencia en cada paso de su proceso de investigación y le ofrece la máxima flexibilidad en su paso de purificación.
Fácil acceso
Es necesario realizar tareas de solución de problemas y mantenimiento de vez en cuando al trabajar con sistemas de cromatografía. Por tanto, se puede acceder fácilmente a todos los tubos y piezas del Sistema de Cromatografía Pure, que permiten al usuario supervisar y hacer un seguimiento del flujo del proceso. Además, el detector UV está conectado externamente a la bomba para que la celda de flujo se pueda limpiar o sustituir fácilmente.
Automatización
La automatización simplifica todo el proceso de separación, ya que permite un funcionamiento seguro y eficiente y reduce cualquier preocupación por acontecimientos impredecibles. El software Pure Essential le guía paso a paso por el proceso de ejecución: cebado, equilibrado, carga de muestras, elución y enjuague. Por último, todos los datos de ejecuciones se guardan automáticamente y están disponibles en cualquier momento mediante informes detallados.

Mas información:

- El Sistema de Cromatografía Pure Essential ofrece como ventajas un diseño y una configuración básicos
- Gracias a las reducidas dimensiones del instrumento, se puede colocar incluso en laboratorios abarrotados, y su solidez lo hace altamente fiable
- Básico a la vez que potente gracias a una bomba de 50 bares, sensores de presión y reguladores
Solución modular y ampliable
Siempre se empieza con lo mínimo. BÜCHI le proporciona asistencia en cada paso de su proceso de investigación y le ofrece la máxima flexibilidad en su paso de purificación. El Sistema de Cromatografía Pure C-900 acelera sus procesos de separación y aumenta la reproducibilidad. No obstante, en cualquier momento puede aumentar aún más la eficiencia de sus procesos de separación añadiendo un colector de fracciones y un detector UV para disfrutar de estas ventajas: recolección de fracciones activada por volumen o UV, supervisión de la eficiencia de los procesos de separación, detección de compuestos durante una ejecución y reducción de la evaporación de disolvente gracias a una recolección más precisa.
- Entre las posibilidades de ampliación se incluye la incorporación de un detector UV y un colector de fracciones al Sistema de Cromatografía Pure C-900
- No hay límites con respecto al tamaño de las fracciones recogidas
- Existen opciones adicionales que le permiten ser flexible con respecto a la carga de muestras (líquidas y sólidas) y el soporte para cartuchos (cualquier tamaño entre 4 – 5.000 g)
Cromatografía Plug and Play
Con los Sistemas de Cromatografía Pure Essential, las separaciones flash son más sencillas que nunca gracias a varias características clave del hardware y el software.
El Sistema de Cromatografía Pure Essential se puede instalar manualmente de manera sencilla, y tanto el colector de fracciones como el detector UV se configuran automáticamente. Gracias al innovador software de última generación, el sistema se puede controlar de manera intuitiva. Con solo unos clics, se puede programar un método o adaptar parámetros sobre la marcha.
- Navegación intuitiva proporcionada por un innovador software de última generación (métodos, datos de ejecuciones y ajustes generales)
- Se puede acceder fácilmente a todos los tubos y piezas del instrumento, que permiten al usuario supervisar y hacer un seguimiento del flujo del proceso
- La automatización simplifica todo el proceso de separación
¿Por qué el Nuevo Sistema de Cromatografía Pure Essential de BÜCHI es ideal si me encuentro dando mis primeros pasos en Cromatografía?
El Sistema de Cromatografía Pure Essential se centra en los aspectos básicos de cualquier aplicación flash básica. Gracias al diseño del instrumento, su modularidad y su facilidad de uso, le resultará muy fácil empezar con la automatización y se beneficiará de una mayor velocidad y de infinitas posibilidades.
Tamaño compacto y solidez
Gracias a las reducidas dimensiones del Sistema de Cromatografía Pure Essential, se puede colocar incluso en laboratorios abarrotados. La bomba, que incluye el detector UV, se puede situar encima del colector de fracciones para ahorrar espacio. El dispositivo ofrece como ventajas una construcción de calidad sólida en general con cabezales de bomba de acero, racks del colector de fracciones extremadamente robustos y un resistente soporte para cartuchos. Está fabricado para resistir los rigores del uso diario.
Básico a la vez que potente y seguro
Aunque es básico, el Sistema de Cromatografía Pure Essential destaca por su potencia y seguridad. La bomba de 50 bares puede gestionar cualquier aplicación flash, con partículas de sílice grandes o pequeñas. Sus tres pistones garantizan un flujo sin pulsaciones que contribuye a una separación eficiente. El detector UV es muy sensible y permite detectar compuestos en concentraciones incluso más bajas. Además, el instrumento está equipado con un sensor de presión y es poco susceptible a los derrames gracias a su diseño inteligente.
Muchas separaciones flash solo requieren una configuración básica. Solo se necesita una bomba para agilizar el flujo de disolventes y muestras y, opcionalmente, un detector UV y un colector de fracciones.
Carga de muestras y recolección de fracciones sin límites
El Sistema de Cromatografía Pure Essential se adapta a sus necesidades.
El colector de fracciones es compatible con recipientes de diferentes tamaños y diseños: tubo, embudos y matraces. Además, el usuario puede elegir entre la carga de muestras líquidas o sólidas en función de la aplicación. Las muestras líquidas se pueden inyectar directamente mediante una válvula de inyección manual o mediante bucles o cámaras de muestras con capacidades de volumen entre 5 y 1.000 ml.
Se adapta a sus aplicaciones
Las aplicaciones pueden variar ampliamente en cuanto a escala y complejidad. Con el Sistema de Cromatografía Pure Essential, tendrá el equipo que necesita para cualquier aplicación. Puede conectar cualquier tipo de cartucho flash (con tamaños entre 4 y 5.000 g) o columnas de vidrio (con un diámetro interno de hasta 100 mm y longitudes de hasta 900 mm) en función de la cantidad de muestra que se vaya a purificar. Se puede instalar una cámara de mezclado a alta presión adicional (con tamaños entre 2,5 y 23 ml) para mezclar correctamente sus disolventes, especialmente cuando se usen en concentraciones bajas.
La separación de una mezcla suele acabar con múltiples fracciones recogidas. La concentración de estas fracciones se puede llevar a cabo mediante el Rotavapor® de BÜCHI o mediante la tecnología Vortex de evaporación con el SyncorePlus de BÜCHI, que permite procesar varias muestras a la vez. El Sistema de Cromatografía Pure Essential es compatible con los mismos tubos de vidrio y racks del Rotavapor® y el SyncorePlus, lo que maximiza la eficiencia de sus procesos.
Siempre se empieza con lo mínimo. BÜCHI le proporciona asistencia en cada paso de su proceso de investigación y le ofrece la máxima flexibilidad en su paso de purificación.
Fácil acceso
Es necesario realizar tareas de solución de problemas y mantenimiento de vez en cuando al trabajar con sistemas de cromatografía. Por tanto, se puede acceder fácilmente a todos los tubos y piezas del Sistema de Cromatografía Pure, que permiten al usuario supervisar y hacer un seguimiento del flujo del proceso. Además, el detector UV está conectado externamente a la bomba para que la celda de flujo se pueda limpiar o sustituir fácilmente.
Automatización
La automatización simplifica todo el proceso de separación, ya que permite un funcionamiento seguro y eficiente y reduce cualquier preocupación por acontecimientos impredecibles. El software Pure Essential le guía paso a paso por el proceso de ejecución: cebado, equilibrado, carga de muestras, elución y enjuague. Por último, todos los datos de ejecuciones se guardan automáticamente y están disponibles en cualquier momento mediante informes detallados.
Mas información:

























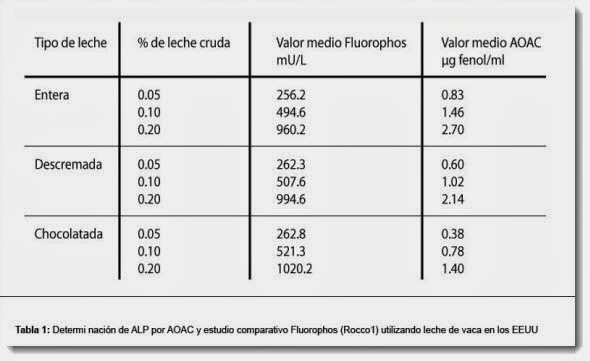
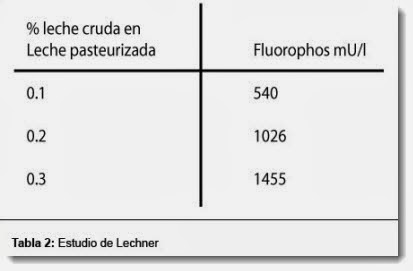























, desvío patrón y coeficiente de variación (CV, %)")
